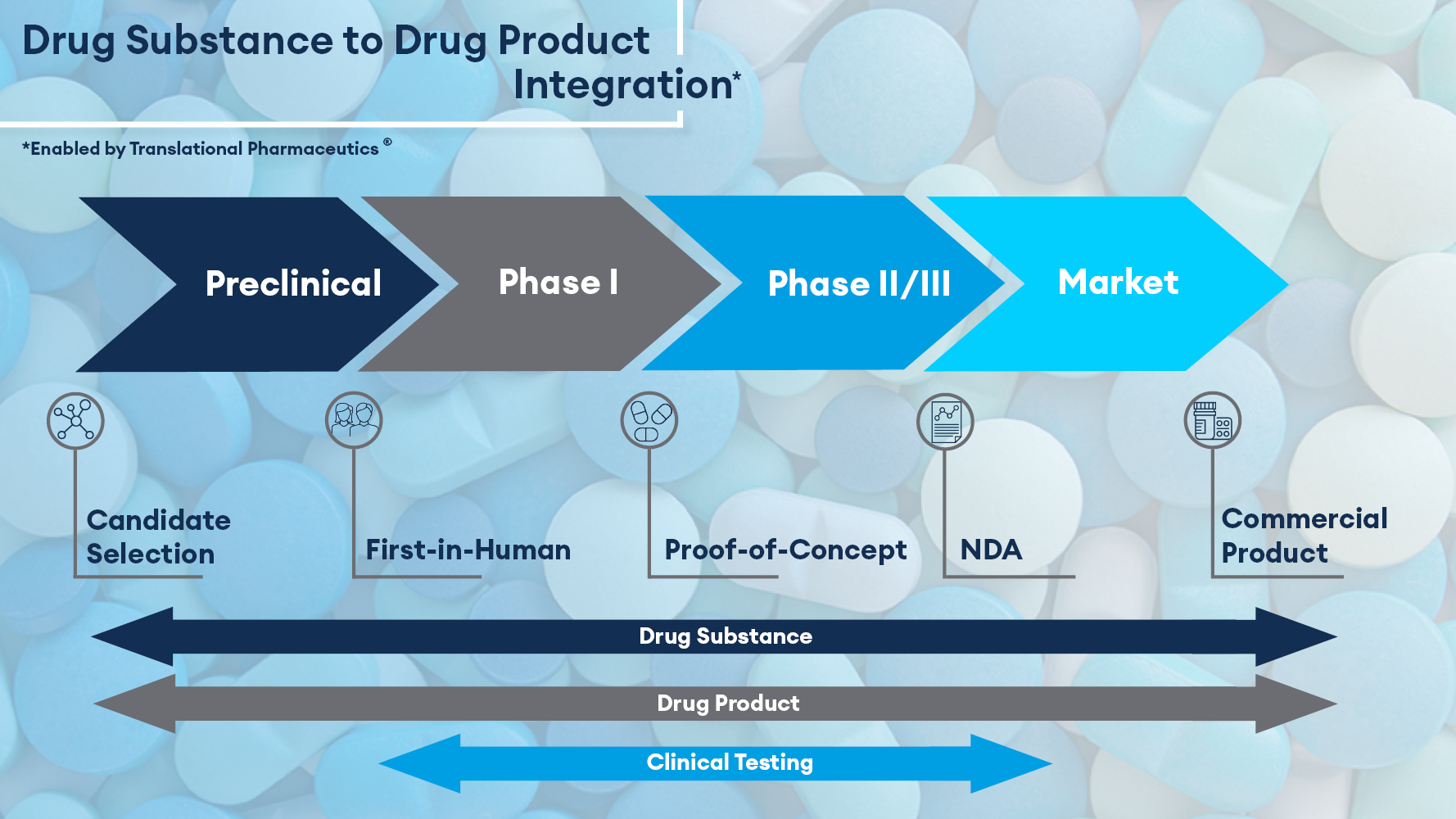
Summary: Dr. Asma Patel shares strategies for accelerating modified-release oral formulation development, focusing on overcoming challenges in achieving target release profiles and bioavailability. She highlights how Translational Pharmaceutics® can be used for modified-release drugs to streamline decision-making, minimize risk, and shorten development timelines.
Oral modified release formulations enable control over the rate and location of a drug’s release in the gastrointestinal (GI) tract to achieve specific therapeutic benefits in comparison to immediate release formulations.
Benefits of modified-release formulations include maintenance of drug plasma levels over a prolonged period to reduce dosing frequency, attenuation of drug peak-to-trough ratios to lower peak-related adverse events (AEs) and improve efficacy, and drug delivery to a particular anatomical site for the treatment of local gastrointestinal (GI) disease.
Drug delivery can be optimized to balance therapeutic needs, by managing AE profiles and reducing dosing frequency, both of which can contribute to improved patient compliance. There are also commercial benefits for modified-release formulations that are prevalent as part of product lifecycle management (LCM). Modest reformulation of an already approved drug from an immediate-release formulation to modified-release format allows both line and patent extension opportunities and continued market exclusivity.
A variety of modified-release technologies are available, eliciting a wide range of control on drug release and drug delivery. Careful selection of appropriate excipients and delivery technologies are key to the design of modified-release formulations fulfilling specific performance requirements, from gastro-retention formulation to a sustained release formulation, as shown in the table below.
While the development of modified-release drugs has historically been a part of late-stage development or LCM strategies, there are increasing examples of where modified-release has been utilized in the development of new chemical entities (NCEs). In all cases, a clear definition of the Target Product Profile (TPP) is important to outline the desired characteristics of the drug product required to deliver the desired in vivo performance. The TPP is based on the drug product requirements including the intended clinical use, dosage strength(s), drug release characteristics, stability, and other product quality criteria.
Many modified-release technologies can be used to control the rate and time of drug release to achieve a particular TPP. A developer is therefore faced with the need to select the strategy that will provide optimal results in the most efficient and cost-effective manner.
| Modified release format | Objective | Formulation technology |
| Gastro-retention | - Keep the formulation in the stomach for an extended period to maximize the duration of absorption or therapeutic activity.
| Swellable tablets (monolithic, bilayer, trilayer) |
| Gastric bypass | - Prevent the release of the drug in the stomach and/or upper gastrointestinal tract.
- Overcome first-pass metabolism or gastric irritation.
| Enteric-coated tablets or capsules |
| Sustained or extended release | - Extend the in vivo release profile of the drug or enable once-daily dosing.
| Matrix tablets, coated tablets, or multiparticulates |
| Targeted or controlled delivery | - Release the drug at or near the intended site of absorption or action.
- Have either immediate or extended-release characteristics.
- Deliver time, pH or microbially-triggered release.
| Tablets, capsules or multiparticulates |
| Biphasic release | - Eliminate the need for repeat dosing.
- Provide rapid therapeutic effect from an immediate release layer and extended dosing via a sustained release layer.
| Bilayer tablets or multiparticulates |
| Pulsatile release | - Release the drug as a pulse after a predetermined lag time — designed according to the body’s circadian rhythm.
- Provide release mechanism beneficial for drugs where time-dependent dosing is required or those that undergo first-pass metabolism.
| Bilayer tablets or multiparticulates |
How is Translational Pharmaceutics® used for modified-release drugs?
Selection of a specific modified-release platform and optimization of the quantitative levels of critical-to-performance excipients in that formulation can be challenging based on surrogate nonclinical, in vitro, or in silico data, and the recognized lack of predictability of these models to performance in humans. Traditional development also means the time and cost of taking multiple options into a clinical PK study can be prohibitive.
The Translational Pharmaceutics® platform is unique to Quotient Sciences, offering integrated development programs with in-study protocol flexibility to enable real-time optimization of key formulation variables based upon arising clinical data. It enables modified-release formulation technology platform(s) to be assessed in the identification of the best technology to achieve the desired TPP.
There are numerous potential formulation strategies available for modified-release dosage forms. Selecting a specific platform and the quantitative levels of critical-to-performance excipients in that formulation can be challenging based on surrogate nonclinical, in vitro, or in silico data.
How is a design space used with Translational Pharmaceutics® to optimize modified-release formulations?
In-study protocol flexibility using Translational Pharmaceutics® can enable the optimization of key variables based on actual clinical data and/or the assessment of multiple technology platforms to achieve the desired TPP. Offering potential benefits in terms of PK variability and bimodal release combination flexibility, could be compared to a matrix modified-release tablet, which could be easier to commercialize if performance was sufficient.
Formulation adjustments within a mapped design space included in the regulatory submission are permissible. Design space methods bracketing several formulation parameters (e.g., drug content, functional excipient content, drug:polymer ratio, surface area volume ratio, and coating composition/thickness) can be used to allow any composition within defined ranges to be selected, made, and dosed.
The design space concept can be applied to any formulation, drug product, or dosage form. The goal in modified-release formulations is to address all the adjustable, critical-to-performance parameters that can influence release rate and PK profile.
Case Study: Development of an optimized modifed-release tablet formulation for initial proof-of-concept trials using Translational Pharmaceutics®
SLx-2101, a novel PDE-5 inhibitor1 was being developed by Surface Logix as an antihypertensive agent. A Phase II pilot clinical study using an IR tablet determined it was necessary to develop a once-daily modified-release formulation to reduce Cmax-related AEs and ensure the 24-hour PK profile remained within the therapeutic window.
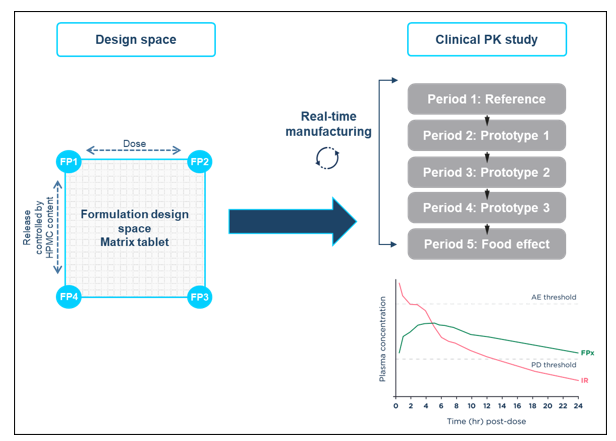
Using formulation design space concepts, a strategy built upon ICH Q8 Development Pharmaceutics, and Quality-by-Design principles, a HPMC-based matrix modified-release tablet formulation was developed for assessment in an adaptive relative bioavailability Phase I study to optimize the modified-release tablet based on human clinical data.
A two-dimensional formulation design space was established covering dose strengths between 10-20 mg and sustained drug release durations between approximately 12 and 20 hours.
The relationship between key formulation variables and formulation performance was investigated. Representative formulations at the extremes and the mid-points of the design space were manufactured and characterized to demonstrate that the performance of the formulation can be controlled by varying the levels of drug loading and HPMC in the formulation.
The SLx-2101 modified-release tablet formulation within the formulation design space was manufactured in real-time and evaluated in a flexible clinical study, avoiding the restriction of only dosing pre-defined formulation compositions. The formulation selection was driven by clinical data from the previous dosing period and the optimal modified-release formulation was identified in 6.5 months.
Summary
Selection of a modified-release platform can be challenging, given the lack of predictive models for human outcomes. The use of formulation design spaces, integrated manufacturing, clinical testing, and flexible clinical protocols can enable the assessment of modified-release platforms to de-risk development, identify the best technology to achieve the desired TPP and thereby maximize the probability of success and reduce development time, getting treatments to patients faster.
References
1. DiMasi J and Wilkinson M. The Financial Benefits of Faster Development Times: Integrated Formulation Development, Real-Time Manufacturing, and Clinical Testing. TIRS, June 2020.
2. USFDA. Conference on Harmonization (ICH) and FDA Guidance for Industry, Q8 (R2) Pharmaceutical Development 2009. https://www.fda.gov/media/71535/download. Accessed May 30, 2019.
3. McDermott J, Scholes P. Formulation design space: a proven approach to maximize flexibility and outcomes within early clinical development. Therapeutic Delivery. 2015;6(11):1269-1278. doi.org/10.4155/tde.15.76.
4. Lin, W, et al. Development of a Formulation Design Space for SLx-2101 Modified Release Tablets to Enable a Flexible Phase I Pharmacokinetic Study (Controlled Release Society Annual Meeting 2010).