


Summary: Kate Darwin, Vice President of Regulatory Affairs at Quotient Sciences, discusses the recovery of MHRA approval times for Phase I trials in the UK. Despite initial delays due to resource shortages, MHRA approval times have improved and consistently meet statutory timelines as of mid-2023. Kate explains the Combined Review process, which streamlines regulatory and ethics reviews into a single application. Quotient Sciences has successfully managed numerous applications under the UK MHRA Combined Review process, ensuring efficient trial approvals and maintaining high standards in clinical research.
*MHRA timelines and data cited were current as of publication date on January 17, 2025
Why the UK remains a great place to do Phase I research
From discussions with clients over the last 6 months, it’s become clear that there are many misconceptions about UK clinical trial review timelines since the introduction of the Combined Review process.
In this blog, we speak with Kate Darwin, Vice President of Regulatory Affairs at Quotient Sciences, who dispels the myths and explains why the UK remains a great place to do Phase I research.
Q: What is Combined Review?
A: In January 2022, the UK introduced Combined Review (CR), a new process for obtaining regulatory approval for clinical trials. The key feature of CR is that an applicant makes a single application and obtains a single opinion from the MHRA and an independent Research Ethics Committee (REC). It’s a much more streamlined process than having separate applications and approvals.
Q: What is the application process under Combined Review? What are the timelines?
A: Applicants manage CR applications via an online system called IRAS (short for the Integrated Research Application System), and upload supporting documents in simple PDF format. Regulatory and ethics reviews are done in parallel. If more information is required, a single, joint request for further information (RFI) is raised by the MHRA and REC, the applicant responds, and a single decision is issued. By law, the initial review must be done within 30 days, and the final outcome must be issued within 60 days.
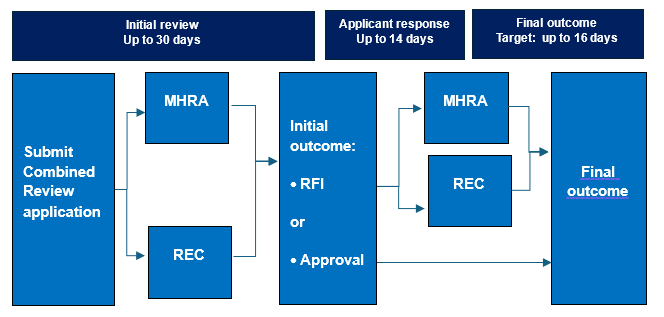
UK Combined Review (CR) takes up to 30 days for the initial review with a final decision issued within a maximum of 60 days from submission.
For trials involving exposure of participants to ionising radiation (e.g. ADME studies), IRAS also generates an application form for Administration of Radioactive Substances Advisory Committee (ARSAC) review.
Substantial amendments are managed via CR and reviewed within 35 days.
Q: There are rumours that UK timelines for trial approval are very long – is that true?
A: No. A few months after the successful launch of CR in 2022, MHRA review timelines did increase, peaking in Q2 2023. The prolonged approval times weren’t caused by CR, but by a shortage of MHRA resources.
In summer 2023, the MHRA made clinical trial authorisations its highest priority, redeployed resources, harnessed external resources and improved processes to clear the backlog.
Since September 2023, the MHRA has consistently met its commitment to meet statutory timelines, and there have been no delays to regulatory approvals for UK clinical trials.
In summary, UK approval timelines have recovered and have been within statutory limits for well over a year.
Q: What is Quotient Sciences’ experience of UK Phase I clinical trial approval timelines under Combined Review?
A: Quotient Sciences’ regulatory team manage CTA applications for over 80% of the clinical trials in our Nottingham, UK clinic, and make about 25% of annual UK Phase I trial applications. We’ve made over 70 successful submissions under CR, and over 30 ARSAC applications in the last 2 years.
Since the decisive action taken by the MHRA in summer 2023, we’ve submitted over 25 clinical trial applications – all were reviewed within 30 days.
Our average approval timelines are shorter than both the statutory 60-day limit and timelines achieved by our sponsors, with some of our approval times as short as 36 days. Our highly experienced Regulatory Affairs team of CMC and clinical experts specialises in early phase applications, and our high volume of submissions allow us to quickly understand and adapt to the latest thinking at the MHRA and RECs and advise sponsors on how to avoid RFIs.
Q: What are current UK timelines for approval of clinical trials and how do they compare internationally?
A: Each month, the MHRA publishes metrics on its performance. The latest metrics show average review timelines (across all clinical trial phases) of 28 days for initial trial applications and 29 days for amendments, with 100% of reviews within statutory timelines.
When comparing UK timelines with other jurisdictions, it’s important to make a fair comparison. For example, the headline FDA timeline for IND review is 30 days; however, unlike UK CTAs, US INDs must be submitted in eCTD published format, which can add 2 weeks to a submission timeline and extend the overall regulatory process.
We’ve increasingly seen sponsors taking a more conservative approach to addition of protocols to open INDs and requesting start-up timelines of up to 90 days to mitigate against FDA questions.
UK approval times compare favourably with those in the EU, where CTA approval timelines, assuming no validation queries, are up to 91 days.4
Q: What are the advantages of doing Phase I trials in the UK?
A: The UK is one of the top destinations for delivery of high-quality commercial early phase clinical trials2, with high scientific, data integrity & ethical standards. We have strong scientific and medical expertise and many highly experienced, MHRA-accredited Phase I units, such as Quotient Sciences’ Nottingham clinic, with a proven track record in delivering internationally accepted trial data, with excellent safety and quality standards overseen by the MHRA. Those units are underpinned by an established Phase I clinical trials infrastructure, including supporting services such as licensed, GMP-compliant manufacturing of high-quality investigational medicinal products, laboratories, monitoring and pharmacovigilance. The attractiveness of the UK business environment is backed up by strong rankings in both the Ease of Doing Business and Global Innovation Index rankings (globally 8th and 4th, respectively)3.
CR provides a straightforward, streamlined application process for clinical trials, with robust regulatory and ethical review, giving sponsors confidence in the quality of their investigational medicinal product, the supporting toxicology, and the scientific integrity and ethics of the trial. Furthermore, the UK’s approachable, respected regulator offers rapid, informal advice and targeted scientific advice. And the UK’s deferral process provides a favourable balance of transparency with commercial protection.
The UK’s robust and pragmatic regulatory environment, coupled with its Phase I infrastructure, enables innovative new medicines to move rapidly from bench to bedside.
Q: Why place UK Phase I clinical trials at Quotient Sciences?
A: Quotient Sciences have been running Phase I clinical trials at our Nottingham clinic for over 30 years. Aspects that set Quotient Sciences apart from our competitors were recently discussed by our Chief Operating Officer and Nottingham, UK site head, Denise Sutton.
In summary, we offer sponsors an accredited, GCP-compliant Phase I clinic that meets the MHRA’s stringent safety and quality standards, with a wide range of pharmacodynamic and safety tests, and integrated GMP manufacturing. The clinic is supported by a multi-disciplinary team, including a large medical team with highly experienced Principal Investigators. We have broad experience across a range of study types, including first-in-human single/multiple ascending doses, human ADME studies, drug-drug interaction studies, ethnic bridging and formulation optimization, and employ adaptive, flexible trial designs. Our real-time electronic data capture system generates GCP-compliant data that meets international ethical and data integrity standards, and is accepted by global regulators, including the FDA and EU authorities. And our high-quality regulatory submissions coupled with our experience in participant recruitment, large volunteer database and volunteer-centric approach ensure rapid recruitment and favourable, reliable trial timelines.
Quotient Sciences have collaborated with our regulators to reposition the UK as a robust, reliable and competitive environment for clinical trials. We’re committed to working with sponsors to ensure the fastest route to clinic.
Get more information about Phase I trials at Quotient Sciences
References
Independent report. Commercial clinical trials in the UK: the Lord O’Shaughnessy review - final report. 26 May 2023 (Commercial clinical trials in the UK: the Lord O’Shaughnessy review - final report - GOV.UK (www.gov.uk))
ABPI. The road to recovery for UK clinical trials, December 2024. The road to recovery for UK industry clinical trials
HM Government Life Sciences Vision, 2021 (Life Sciences Vision (publishing.service.gov.uk))
EMA. CTIS Evaluation Timelines. CTIS training programme, version 2.1, September 2024.