Summary: In this Q&A, Jane McGuffog, Director of Modeling & Simulation at Quotient Sciences, discusses how in silico modeling and simulation (M&S) drive smarter, faster drug development. Jane explains the use of PBPK and PBBM modeling to predict drug behavior, optimize formulations, and support regulatory submissions.
In silico modeling is a proven scientific approach used to inform key development decisions, and can be employed throughout the lifecycle of a drug. In this Q&A with Jane McGuffog, Director of Modeling & Simulation at Quotient Sciences, she explains the applications of M&S in clinical research and developing effective drug products.
Tell us about your background. What drew you to modeling & simulation?
I have worked in drug discovery and development for over 30 years, with a background in DMPK in both small biotech and large pharma. During that time, I have led a number of clinical stage projects (FIH /Phase I/Phase II), encountering all the usual challenges with drug substance and drug product, and predicting human pharmacokinetics.
Experiencing the impact of modeling first-hand, I was lucky enough to be able to apply M&S to several projects via my own team and through outsourcing to partner companies. I am convinced that it really helps make better development decisions, but it is critical to find the right partner if you are looking to outsource these services.
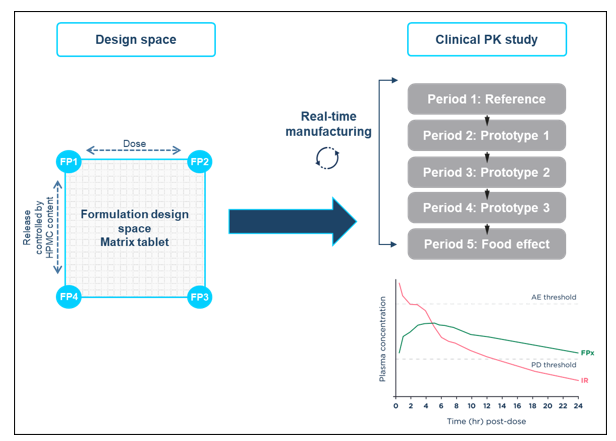
As a client, I worked with Quotient Sciences on two projects. I was particularly impressed with how the Translational Pharmaceutics® platform was applied for drug product optimization for a modified-release product, as a RapidFACT® program. The knowledgeable and collaborative modeling team were a pleasure to work with.
In fact, I joined Quotient Sciences in part because I had such a good experience working with the team! I am incredibly lucky to lead such a talented and experienced group of modelers. They love problem solving and that comes through in everything they do.
How do Quotient Sciences use modeling & simulation in drug development?
Physiologically based pharmacokinetic (PBPK) modeling integrates knowledge of a drug’s characteristics with physiological information to provide important insight into how new drugs will behave in vivo, allowing decisions around drug development to be made confidently. We use it to build models that predict the plasma profiles of drugs.
PBBM modeling builds on PBPK by focusing on how a drug is absorbed. It looks at how things like solubility and how fast a drug dissolves affect its performance in the body. Using these models throughout a drug’s development can save time and money, and lead to smarter strategies.
Physiologically based biopharmaceutics modeling (PBBM) builds on PBPK and focuses on drug absorption, especially how drug biopharmaceutic properties including solubility and permeability, and formulation can affect in vivo performance.
The resultant models can be used to simulate potential in vivo performance of as yet untested formulations and treatment regimes, with corresponding time and budget efficiencies, whilst simultaneously driving better development strategies.
How are PBPK and PBBM used as molecules enter clinical research?
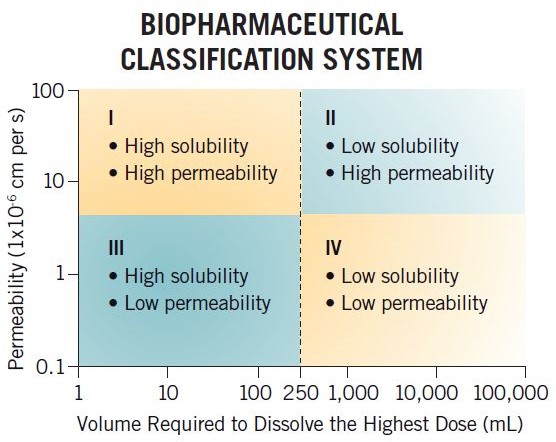
PBPK modeling and PBBM can be used to assess compounds and guide formulation strategies for clinical assessment. By understanding the solubility and permeability of a compound, and an estimation of intended dose range, M&S can enable rational decisions to be made, such as:
- Could micronization of my active pharmaceutical ingredient (API) help increase Cmax/exposure?
- Is precipitation a risk for my compound?
- Will I need to use an enabled formulation to get the exposure I need?
Although there may not be a wealth of data in early development, it is possible to make initial risk assessments on a limited dataset. For example, M&S can use in-silico predictions from chemical structure to assess small intestinal permeability.
Teaming up M&S with our preformulation team is another option for clients looking to move efficiently and cost effectively from fit for purpose preclinical formulations to something fit for FIH , and looking forward to formulation development towards your value inflection point.
How can PBPK and PBBM M&S be used in early development?
During clinical and product development, M&S can provide insight to help answer key questions and guide the development strategy before the drug has even been dosed to a human, such as:
- Can I change my API particle size without significantly impacting pharmacokinetic (PK) parameters, like Cmax and AUC?
- How much will my drug’s PK parameters change if my drug is taken with a meal?
- What dose can I give to an older person or child to obtain equivalent exposure to a healthy adult?
- Can I use my in vitro data to assess DDI risk?
Although it is possible under certain circumstances to use M&S data to secure biowaivers in lieu of clinical studies, in most cases it is not a replacement for a clinical trial, though it can often reduce the number of clinical studies that might be needed.
Instead, M&S maximizes data from in-vitro, clinical, and pre-clinical studies to aid the understanding of a drug in the human body. From this, appropriate formulation design and development strategies can be made.
How can PBPK and PBBM M&S be used in late development?
In later development, modeling is increasingly used to support chemistry, manufacturing, and controls (CMC) aspects of regulatory applications.
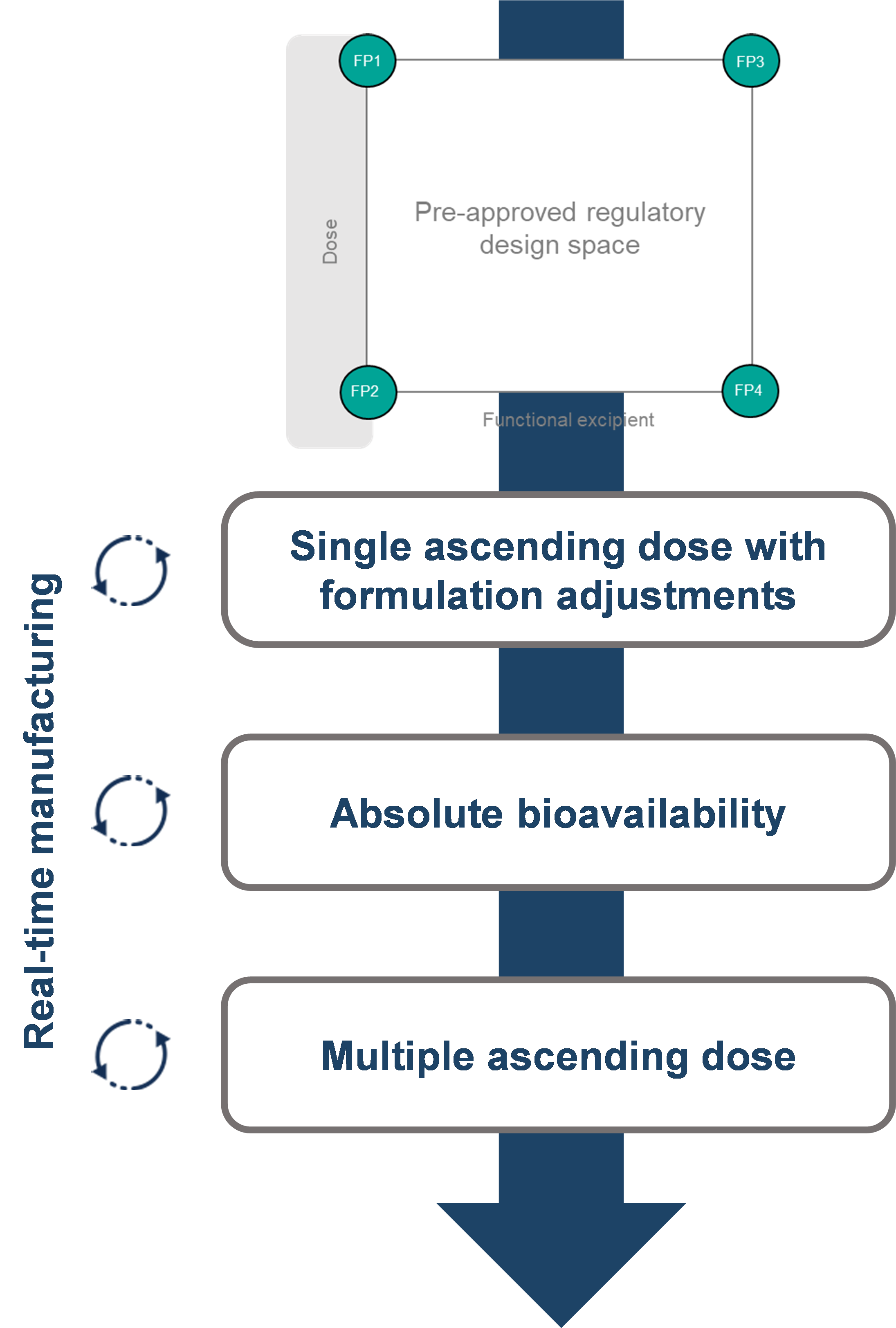
PBBM can help define product specifications (particle size and dissolution rate), that are linked directly to clinical performance. This supports regulatory acceptance and reduces the need for extensive clinical trials.
It can support Virtual Bioequivalence (BE) studies, enabling ‘safe space’ for formulation changes. In certain cases, this can justify biowaivers and reduce the need for clinical bridging studies.
Also, PBBM can be used to perform risk assessment for manufacturing changes, such as excipient or process changes, predicting their impact on bioavailability. It can be involved in lifecycle management, supporting decisions throughout the product lifecycle, including reformulation, scale-up, and global registration
How does your team collaborate with other departments at Quotient Sciences to achieve project goals?
The M&S team operates within Quotient’s Consulting team which brings together a wide range of scientific disciplines into a single group.
All programs start with a scientific discussion to understand the development challenges at hand and form the questions that M&S can look to help answer. We have worked with clients who have had little experience of PBBM, as well as those who are very experienced modelers.
M&S involves integrating information from many disciplines, so a large knowledge base is required. In addition to having team members with varied backgrounds—from chemistry, pharmacokinetics, and biopharmaceutics to mathematics and coding—we work cross-functionally to access the knowledge that Quotient Sciences has built over more than three decades. The team can connect you with other expertise within Quotient Sciences, too.
Whether we are doing a stand-alone M&S program or working as part of an integrated project, we work collaboratively and flexibly, and are always client-focused.
What software does Quotient Sciences use for M&S?
Most of the modeling work performed at Quotient Sciences uses GastroPlus™ software to build virtual models to describe mechanistically the behavior of a drug in the body.
GastroPlus™ mechanistically describes not only drug dissolution (like location, rate, and precipitation potential) but also drug absorption (such as rate, intestinal location, and affinity for metabolic gut enzymes and transporters), tissue and blood distribution, and finally excretion (metabolic and/or renal clearance/other mechanism).
The team also have experience using the open source platforms PK-Sim® and MoBi®, which provides us with flexibility and allows us to select the right tool for the right job.
Do you do any other types of M&S work other than PBPK modeling?
The M&S group is also experienced in developing numerical (empirical rather than mechanistic modeling) in-vitro/in-vivo correlations (IVIVCs), which allow a relationship between in-vitro performance (dissolution) and in-vivo outcome (PK parameters Cmax and AUC) to be developed. A successful Level A IVIVC can be used to inform drug product development or used as part of a regulatory submission.
In addition, the group have experience in drug drug interaction (DDI) modeling to assess potential DDIs, inform clinical study design, and support regulatory submission. A poster was presented by Kevser Sevim at the 2024 American Association of Pharmaceutical Scientists (AAPS) PharmSci 360 conference, showcasing the development and validation of a PBPK model to simulate drug-drug interactions (DDIs) involving belumosudil.
I’ve been impressed by the team’s innovative modeling approaches, particularly in the area of oral peptide delivery using permeability enhancers. Ricardo Diaz de Leon Ortega has led this work and presented recent advancements at several key events, including our co-sponsored workshop at the Controlled Release Society Annual Meeting in Philadelphia, the 16th PharmSci APS 2025, and PAGE in Thessaloniki, where he was joined by Dannielle Ravenhill to share insights from Quotient Sciences’ Modelling & Simulation team.
For more information about Quotient Sciences’ M&S capabilities, click here or contact us directly here.