At Quotient Sciences, our focus is on bringing lifesaving therapeutics to patients quickly. We work with more than 200 pharmaceutical and biotech companies across disease indications, and one area gaining increasing attention is the development of orphan drugs for rare diseases.
An orphan drug is a drug for a rare disease or condition, affecting a small percentage of the population (less than 200,000 people in the US, and not more than 1 in 5000 in the EU). Worldwide there are over 300 million people living with one or more identified rare diseases representing 3.5% - 5.9% of the global population. Some rare disease treatments have been “orphaned” or discontinued because there was not enough financial incentive to continue development or production.
To encourage industry R&D the Orphan Drug Act by the FDA incentivizes drug development for rare diseases, which was reciprocated in Europe in 1999 via the Orphan Drug Regulation. These regulatory frameworks recognized the importance of developing new treatments to address these unmet clinical needs.
Can you provide a little bit of background on this important area of drug development?
Global regulatory agencies have sought to provide an impetus to pharma and biotech organizations for the development of new therapeutics for orphan diseases by offering enhanced regulatory support, expedited review times, reduced submission fees, and market exclusivity periods. We have seen the benefits of these incentives most prominently in the US, with over 50% of NDA approvals in the last two years being for rare diseases. In Europe of the 66 new medicines authorized by EMA in 2019, 7 of these (~10%) were orphan drugs. From a global perspective, it is estimated that approximately 30% of new drugs in development today are focused on these therapeutic areas. This is a great success story, but much more still needs to be done to address global patient needs.
What are some challenges that drug developers are still facing when developing these therapeutics?
There are four major CMC challenges that we have seen in the orphan drug space recently, which are arguably exacerbated by regulators providing pathways for expedited development. These include the effective development of patient-centric dosage forms based on molecule properties and patient needs, particularly given many rare diseases will be in pediatric populations. In addition, there are challenges to quickly identify optimized drug products, whose performance is preferably demonstrated in humans, prior to initiating protracted, difficult-to-recruit patient trials. Linked to this are then the challenges of implementing a tailored manufacturing and supply plan for drug products into these patient trials, and then finally finding a long-term partner for the scale-up and commercial manufacturing of what will inevitably be low-volume products.
How can Quotient Sciences help address those challenges that you mentioned above?
In the orphan drug space, Quotient is keenly aware of the need to “start with the end in mind”. Our approach has been to prioritize the key API characterization data required, which allows our scientific experts to recommend a selection of the appropriate API form as well as inform a data-driven strategy for preclinical and clinical pharmaceutical development.
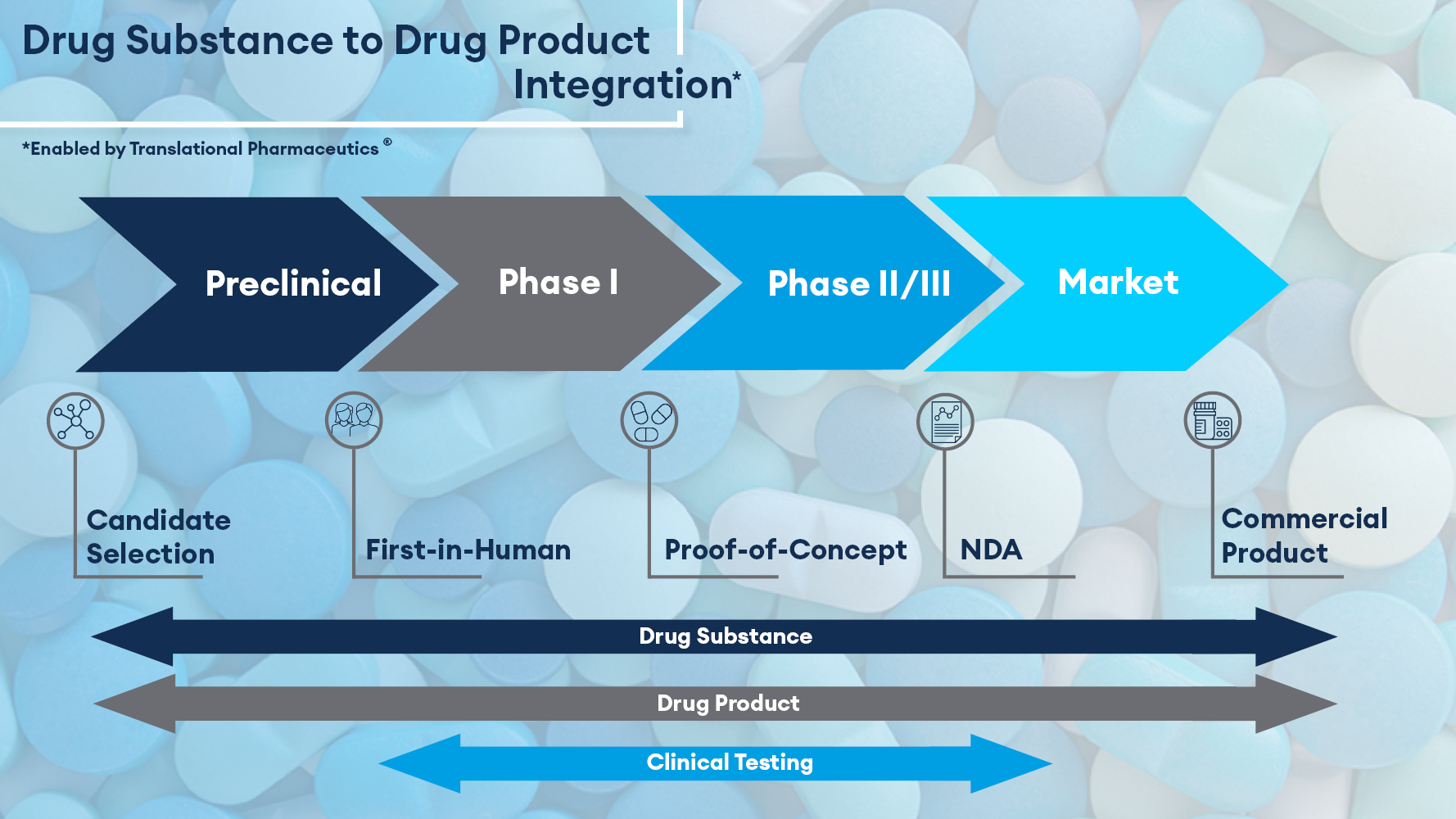
At Quotient Sciences, we have demonstrated the benefits of embedding formulation flexibility within FIH trials to enable a “patient-ready” formulation to be identified and be ready for POC studies in an average of 12 months, less than half the time of the industry standard. We accomplish this by integrating real-time manufacturing and clinical testing. Studies can start quickly with a simple FIH formulation, allowing parallel development of a solid oral format, which is introduced into a later part of the FIH protocol without the need for a separate clinical PK bridging study. At the end of dosing healthy volunteers in the FIH trial, the lead formulation is manufactured and supplied to the patient POC study. No further pharmaceutical development or clinical bridging work is required. This is ideally suited to rare disease therapeutics, as patient trials can be initiated with confidence in the clinical performance of the drug product.
We have also developed significant expertise in providing manufacturing and supply chain solutions for challenging patient trials for over a decade. We are aware of the difficulties presented where recruitment rates are sporadic across multiple study sites in multiple countries. There may also be a need to customize the drug product based on specific patient attributes. To that end, we are able to support complex clinical trials based on program needs, whether with “traditional” large batch manufacturing, all the way through to the personalized product manufacture. Our focus is on helping the customer get the right product to the right patient at the right time.
Looking ahead, modest market potential will mean an even greater focus on managing program time, cost, and risk. Lean and flexible programs will be imperative to progress molecules effectively and efficiently from candidate selection through to commercialization. By definition, access to patient populations during clinical research will be challenging and may require increasing personalization of the drug product based on individual patient needs. A flexible manufacturing and supply platform will be key. Finding the right commercial manufacturing partner will be challenging given the relatively low production volumes required and the high variability in product formats and configurations.